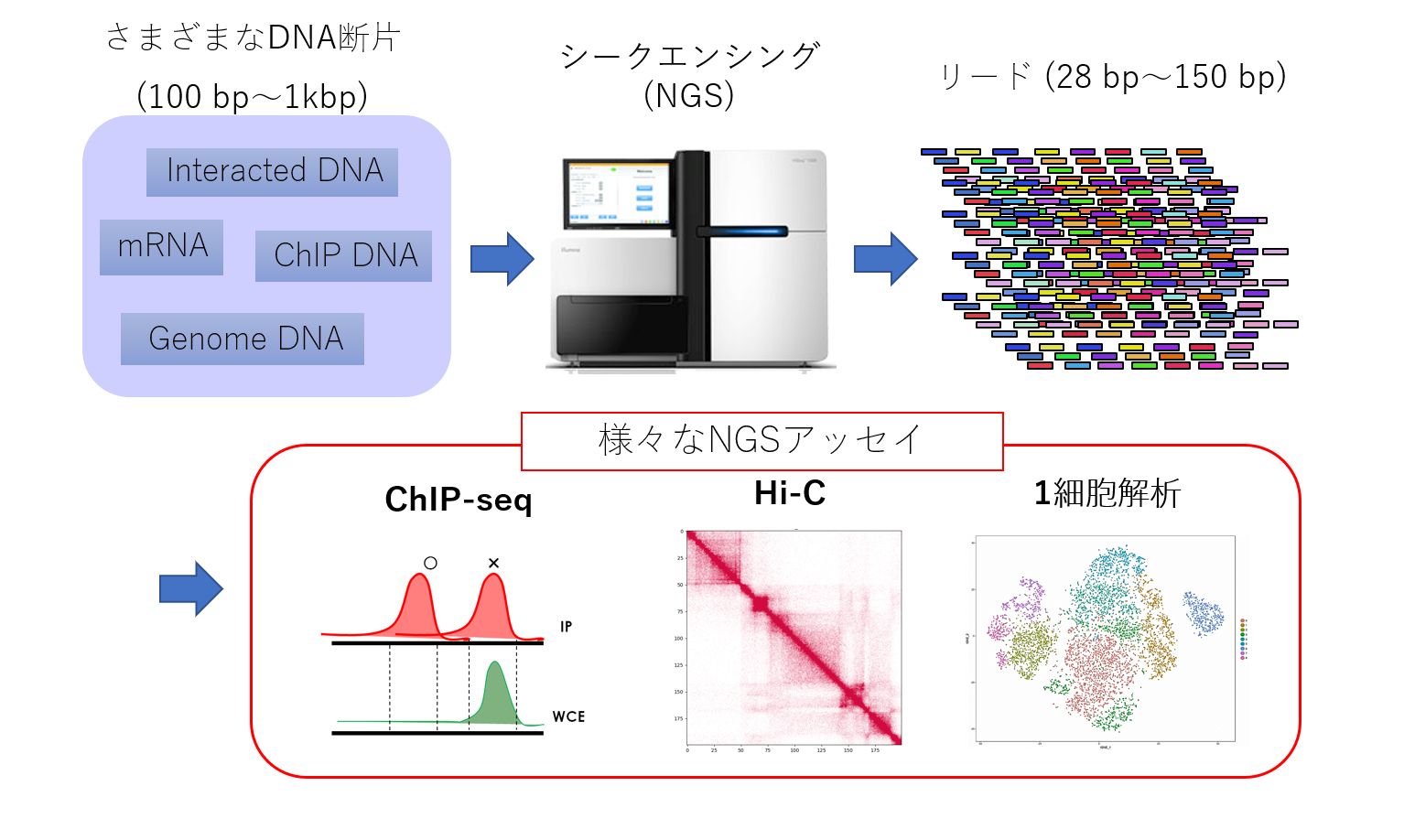
研究概要:次世代シーケンサを用いたデータ駆動型ゲノム解析
ゲノム (genome) は「世代を超えて受け継がれる生命の設計図」です。生命活動を維持するための全ての遺伝子はゲノム上に存在します。皮膚や脳,消化管,骨などの臓器を構成する様々な細胞種はその組織に必要な遺伝子群を適切に発現させていますが、その発現を制御するための機能領域(エンハンサーなど)もやはりゲノム上に存在しています。この機能領域のゲノム修飾状態(エピゲノム; epigenome)が変化することが遺伝子発現の「スイッチ」の役割を果たします。ゲノム配列は全身の細胞で同一ですが、エピゲノムは組織ごとに異なるパターンを示し、それぞれの組織が正しく機能するために重要な役割を果たします。データ量にしてDVD1枚分にも満たないヒトゲノム配列の中に、生命のあらゆる情報が詰め込まれているのです。
ゲノム配列には個人差があり、その違いによって、お酒の強さが変わったり、目の色や髪の色が変わったりします。
また、ゲノムが正常に機能しなくなるような問題のあるゲノム変異やエピゲノムの異常が起きると、
がんをはじめとするさまざまな疾患を引き起こしうることがこれまでの研究でわかってきました。
ゲノム上のどこに生命活動に重要な領域があり、どこにどのような変異が入るとどのような影響があるのか、という具体的なメカニズムを知るための学問を「ゲノム学 (genomics)」また「エピゲノム学 (epigenomics)」と呼びます。ゲノム学では、疾患に重要な遺伝子やゲノム領域を見つけ出し、新薬開発などに結び付ける研究(ゲノム創薬)や、より多収量で病害に強い作物への品種改良などの研究、生命の本質的な理解につながる新発見が期待されています。
我々の研究室はゲノム学、特にエピゲノム学をテーマにしています。
次世代シーケンサ(Next Generation Sequencer; NGS, 上図)を利用した種々の解析技術の発展により、遺伝子の転写レベルや、タンパク-DNA結合、DNAメチル化、ゲノム複製、ゲノム立体構造など、さまざまなゲノム・エピゲノム情報を全ゲノム的にとらえることが可能になりました。NGS解析を用いることで、たとえば疾患患者と健常者の細胞を比較して「疾患に重要な遺伝子やゲノム領域」を同定したり、皮膚・脳・筋肉など様々な組織のゲノム情報を網羅的に比較することで「組織ごとに異なる(組織特異的と呼びます)エピゲノム情報とその重要性」を推定することができます。
特に近年は、利用可能なゲノム・エピゲノムデータが急速に増大していることに伴い、大量のNGSデータを一挙に解析し、これまでの常識を覆すような大きな発見をする「データ駆動型大規模ゲノム解析」の期待が急速に高まっています。
既存知識に基づき立てられた作業仮説を実験で検証する「仮説検証型解析」に対し、データ駆動型解析は既存知識に頼ることなく、データそのものに含まれる特徴を利用して新しい発見をする方法です。大量のデータを横断的・網羅的に解析することで、たとえば「あるタンパク質が特定の組織で予想外のタンパク質と相互作用しており、その相互作用が失われることである種の疾患が発症する」というような複雑かつ予想外なメカニズムを明らかにできる可能性があります。
データ駆動型ゲノム解析の課題
それでは、大量のNGSデータから生物学的に重要な情報を得るにはどうすればよいのでしょうか?NGSデータは1サンプルでも既に全ゲノムレベルの情報量を持っており、それが数百、数千サンプルとなるとそれこそ途方もないデータ量になります。また、得られるデータの構造やデータの特性は実験法ごとにばらばらですし、技術的に難しい実験のデータは品質に大きなばらつきが生じます。「ゲノムのどこに着目すればよいのか」が未知であることが多いため、深層学習を用いた画像認識のような教師あり学習を用いることも困難です。
玉石混交のビッグデータから信頼性高く重要な知見を得ることは現在でも非常に難しく、大きな需要があるにも関わらず、大変高いスキルを持った一部の研究者しかそのような解析ができないという現状があります。
レストランに例えるなら、様々な食材(データ)は充実してきたけれど、料理するための下ごしらえ(データ整形)は全くされておらず、料理するための器具(ツール)や、料理できるシェフ(解析者)が足りてない、というような状況です。
上記の課題を克服すべく、我々の研究室ではデータ駆動型大規模ゲノム解析のための技術開発に取り組んでいます。AMED-PRIME「頑健なデータ駆動型エピゲノム解析システムの構築」や創発的研究「大規模ゲノム解析の新たな価値を拓く情報解析基盤の創出」などのプロジェクトを通じ、エピゲノム(ChIP-seq)、遺伝子発現(RNA-seq)、ゲノム立体構造(Hi-C)、1細胞解析(single-cell)などを対象に、種々の新規手法を開発しながら、複数のオミクスデータからなる多数のデータを統合的に解析する「大規模マルチオミクス解析」のためのパイプラインを構築しています(下図)。アルゴリズム面での新規性だけではなく、いかに重要な知見獲得に結びつけるか、という点にこだわりながら研究を進めています。
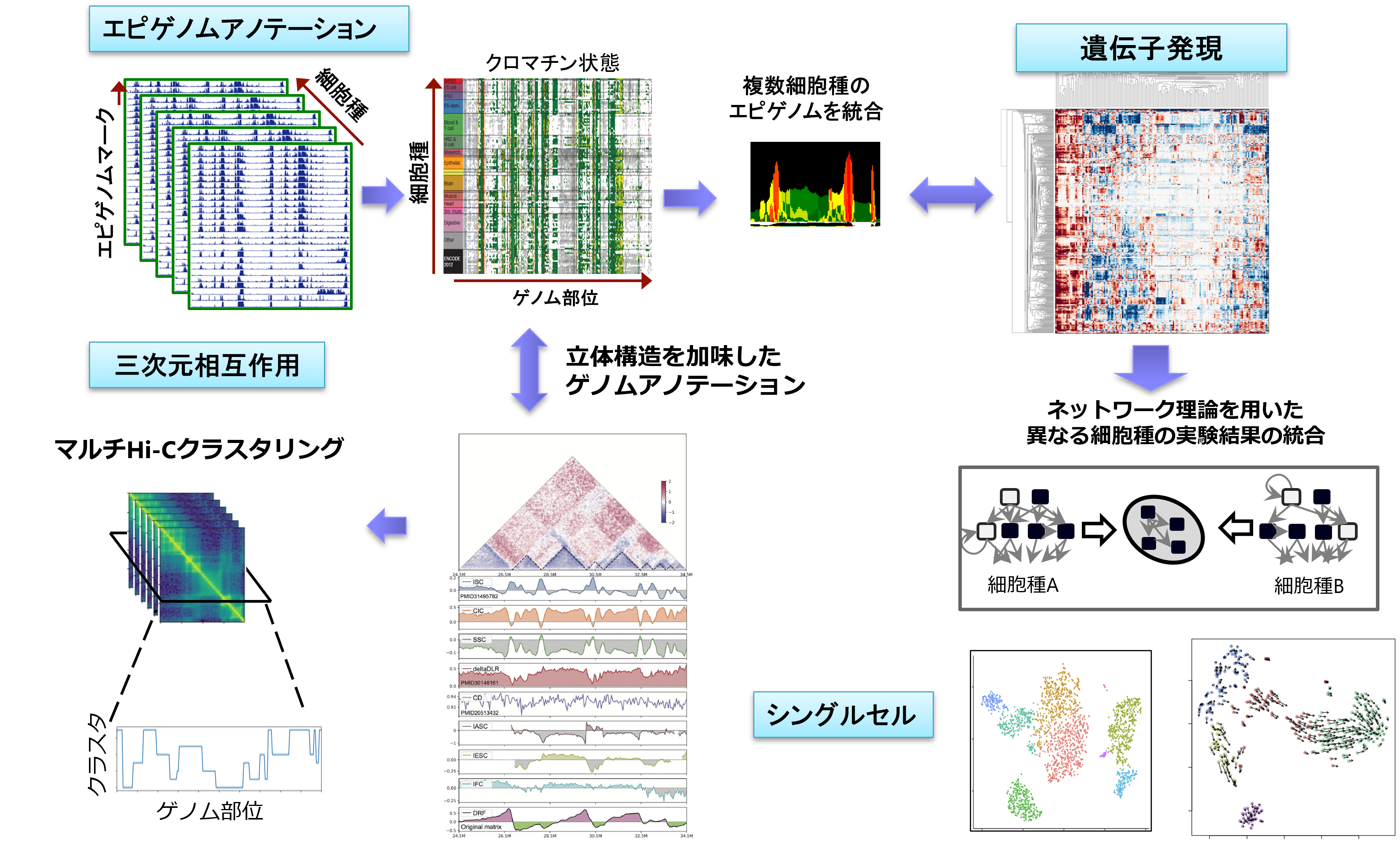
図: データ駆動形ゲノム解析システムの概要
研究概要(専門家向け)
中戸研究室では、計算ゲノム学(Computational genomics)を扱います。様々な実験手法、情報解析手法を用いて、未だ明らかになっていないゲノムの謎を解き明かすことが目標です。解くことが難しい問題をいかに精度良く・効率的に解くかという「情報学」と、まだ世界で誰も知らない新たな知見を色々な実験で明らかにするという「生命科学」の両方の側面を持っていることが、われわれの研究分野の大きな特徴のひとつです。
たとえば、シングルセルデータを用いて遺伝子共発現ネットワークを推定する研究に興味があるとしましょう。「より高精度にネットワークを推定する方法はないかな?」「共発現ネットワークには枝の向き(因果関係)が含まれていないが、因果関係も推定することはできるだろうか?」というような問いが情報学(Bioinformatics)です。これに対し、「得られたネットワークはサンプルのどのような機能・性質を反映しているのか?」「幹細胞のシングルセルデータと分化刺激後のデータからネットワークをそれぞれ推定し、ネットワーク比較することで、細胞分化に重要な新規の遺伝子を発見できるのでは?」というような問いが生命科学(Computational biology)になります。中戸研ではどちらの研究も可能です。
情報学の観点から
現在我々は特に、深層学習モデル、特に近年発展が著しい大規模言語モデルを用いたゲノム配列からのゲノム情報予測に力を入れています。
また、グラフ理論を用いたマルチモーダルデータの統合解析、未知の因子間相互作用予測も新しい研究テーマの柱として取り組んでいます。
テーマの例
・大規模言語モデルを用いたオミクス予測モデルの開発:自然言語処理の分野で大きな成功を収めている大規模言語モデルを活用し、ゲノム配列から様々なオミクス情報を予測する研究です。我々は特に、近年発表されたBorzoiやAlphaGenomeのようなモデルに対して新規のデータセットでファインチューニングを施し、より様々な予測を可能にすることを目指しています。
・ネットワーク理論を用いたマルチモーダル統合解析:遺伝子発現制御機構、タンパク質間相互作用、クロマチンループなどの情報は、節と枝から構成される有向・無向ネットワークとして表現することが可能です。種々のネットワーク理論を用いて複数のネットワークを統合することで、マルチモーダル情報を統合する手法を開発します。
生命科学の観点から
現在我々は特に、エピゲノム(ChIP-seq)、ゲノム立体構造(Hi-C, Micro-C)と遺伝子発現状態(RNA-seq)の関係性に興味を持っており、それらが疾患や細胞分化過程においてどのように変動するのかについて知りたいと考えています。また、シングルセルデータ(scRNA-seq, scATAC-seq)を用いた細胞分化軌道解析、遺伝子ネットワーク解析にも注力しています。
テーマの例
・ゲノム立体構造データと遺伝子発現の関係、コヒーシンの機能解析:ゲノムの立体構造はエピゲノムや転写制御に重要と言われており、そこでは「コヒーシン」というタンパク質が重要な役割を果たしています。コヒーシンの機能は多岐にわたり、その詳細は未解明な点がたくさんあります。コヒーシンのノックダウン実験やコヒーシン病患者細胞の解析を通して、立体構造の重要性、コヒーシンによる制御メカニズムを解明します。
・シングルセルデータを入力とした細胞分化軌道解析と空間解析:シングルセルデータは様々な活用法がありますが、我々は特に、幹細胞からの分化系譜を捉える細胞分化軌道解析、空間情報を保持したまま解析を行う空間オミクスに興味を持ち、新規技術の開発と知見獲得を行っています。
これまでのプロジェクト
大規模言語モデルを用いたオミクス予測モデルの開発
大量に蓄積されている公的エピゲノムデータを学習した深層学習モデルを構築し、エピゲノム情報から重要な特徴を抽出したり、関連するデータを用いて計算機上で疑似エピゲノムデータを生成するデータ補完などのアプローチに取り組んでいます。
遺伝子調節と細胞のアイデンティティーの基本となるクロマチン状態は、ヒストンの翻訳後修飾のユニークな組み合わせによって定義されます。その重要性にもかかわらず、主要な生物学的機能に関する洞察を提供できるクロマチン状態シーケンス内の包括的なパターンはほとんど調査されていません。私たちは、クロマチン状態アノテーションの明確なパターン(クロマチンモチーフ)を検出するBERTモデルであるChromBERTを開発しました。BERT モデルを Dynamic Time Warping (DTW) を使用した motif アラインメントと組み合わせることで、ChromBERT は転写およびエンハンサーサイトと通常関連するクロマチンモチーフを分析するモデルの能力を強化します。
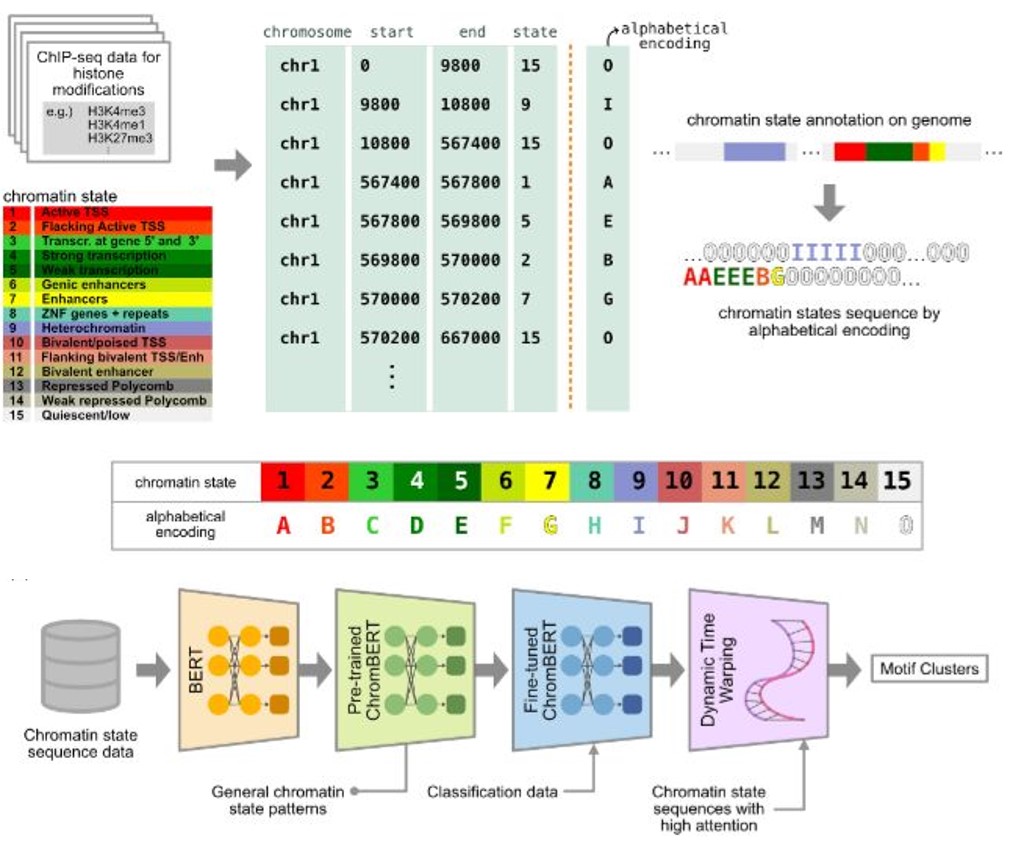
図: ChromBERTの概要
マルチオミクスデータを用いたデータ駆動的ゲノムアノテーション
ヒトゲノムにおいてタンパク質をコードする遺伝子領域はわずか3~5%しか存在しませんが、残りの「非コード領域」においても、遺伝子制御やその他のゲノム機能において重要な領域が多く含まれていることが近年わかってきました。我々は大量のエピゲノム・立体構造データを用いて事前知識を用いることなくデータ駆動的にこれまでに明らかにされていないゲノム上の機能領域を同定することを目指しています。
Hi-C解析ではゲノム立体構造を全ゲノム的に得ることができますが、従来のサンプル間比較はシンプルな可視化やコンパートメント構造の比較にとどまっていました。我々は多数のHi-Cサンプルを横断的に解析し、その情報をもとにゲノム領域をクラスタリングすることが可能な新規手法CustardPyを開発しました。立体構造に重要であることが知られている因子群(コヒーシン・CTCFなど)を欠失させたサンプルから得られたHi-Cデータ群にCustardPyを適用し、TADの境界部分は異なる因子によって制御される複数のサブグループがあること、コヒーシンの機能喪失によりTAD間の立体相互作用がエピゲノム依存的に変動すること、コンパートメントA領域とB領域ではコヒーシンのゲノム上の存在量が大きく異なることなどを明らかにしました。
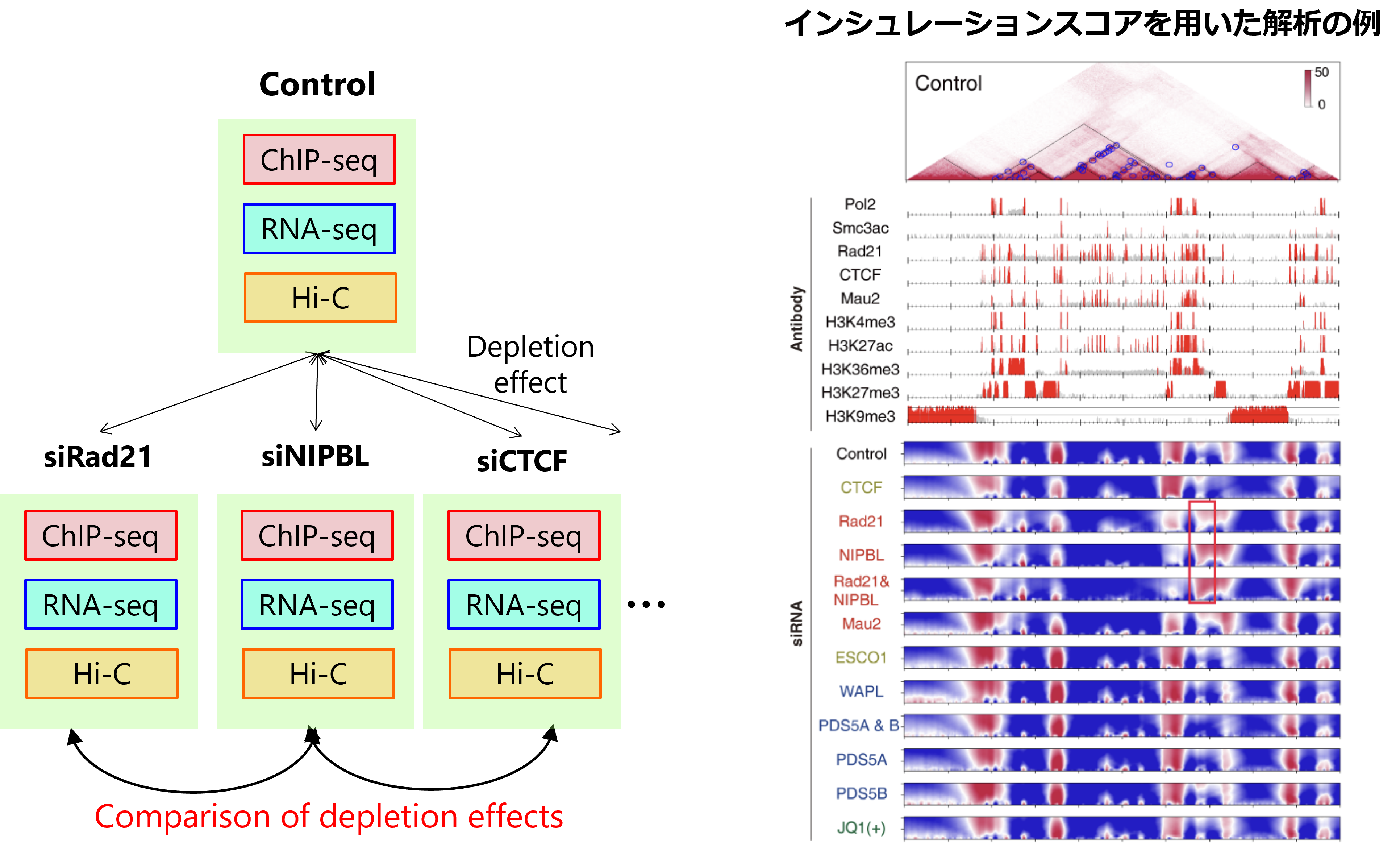
図: 左:立体構造に関わる因子をノックダウンした株を多数作成し、マルチオミクスデータを生成しました。右:クラスタリングによって得られた重要なゲノム領域の例。上:Hi-Cデータの可視化。転写因子及びヒストン修飾の分布。下: Hi-Cデータから計算したMulti-scale insulation score。赤い部分がTADの境界を示す。
コヒーシンはTADの制御、エンハンサー・プロモーターループの媒介、絶縁、RNAポリメラーゼIIの伸長 (elongation) など、様々な機能を介して遺伝子制御に関与していると言われています。コヒーシン(またはその関連因子)の変異は先天性の発達症候群である「コルネリア・デ・ランゲ症候群(CdLS)」や急性白血病など複数のがんの要因となることが知られていますが、具体的にコヒーシンのどの機能がこれらの疾患に関与しているのか、そもそも何故コヒーシンはゲノム上でそのように多岐に渡る機能を同時に果たせるのか、数多くの研究にも関わらず依然として謎に包まれています。
我々は、ゲノム中で数万箇所におよぶコヒーシン結合部位の中から、遺伝子の転写活性化に伴い結合が失われる、すなわち「転写活性と負の相関を示す」ごく少数の遺伝子内コヒーシン結合部位およびクロマチンループを見出し、これを "Decreased intragenic cohesin sites (DICs)"と名付けました。DICの特徴を明らかにするため、ChIP-seq, RNA-seq, Hi-C, ChIA-PETなどから成る計100サンプル以上のゲノムデータを用いた大規模なマルチオミクス解析を実施しました。これにより、コヒーシンの転写制御機構としてこれまで報告されていたエンハンサー・プロモーター相互作用などに加えて、RNAポリメラーゼIIの滞留または伸長阻害という転写を負に制御する機構を持つらしいこと、その機能がコヒーシン病患者でも変動しているらしいことなどを報告しました。
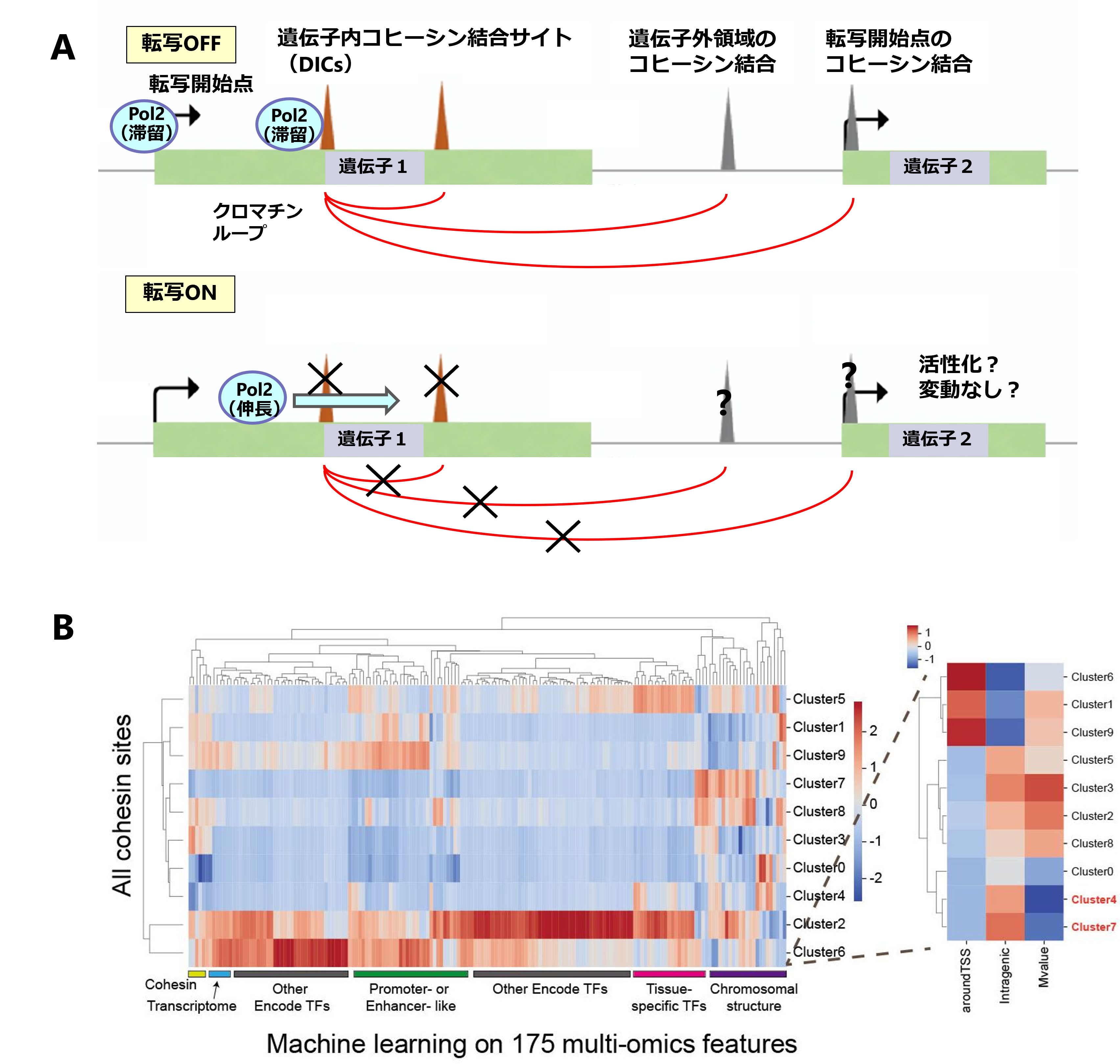
図: A: 遺伝子内領域 (intragenic regions)に存在するコヒーシン結合サイトのうち、転写活性化と共に結合が失われるコヒーシン結合部位をDICsと定義しました。B: 大規模なゲノムデータセットと機械学習を組み合わせた解析により、DICsに関連する多数の特徴を抽出しました。
ネットワーク理論を用いた遺伝子相互作用解析
遺伝子制御ネットワークは遺伝子発現制御機構を有向グラフで表現し、共発現ネットワークは様々な細胞種における遺伝子の共発現・排他的発現パターンを符号付き無向グラフで表現します。また、タンパク質間相互作用のタンパク質ネットワークもよく用いられるネットワークのひとつです。これらの異なるモダリティを持つ異種ネットワークを統合的に解析し、未知の知見を得る解析手法の開発に取り組んでいます。
疎なscRNA-seqデータから遺伝子共発現ネットワークを頑健に推定する手法"EEISP"を開発しました。遺伝子単位ではなくネットワーク単位でサンプルを比較することにより、従来の遺伝子発現変動解析では得られなかった新規のマーカー遺伝子候補を同定することが可能になります。本手法をヒト膠芽腫幹細胞データに適用し、幹細胞・非幹細胞間で遺伝子ネットワークの比較解析を実施した結果、新規の膠芽腫幹細胞マーカー遺伝子候補を複数同定しました。
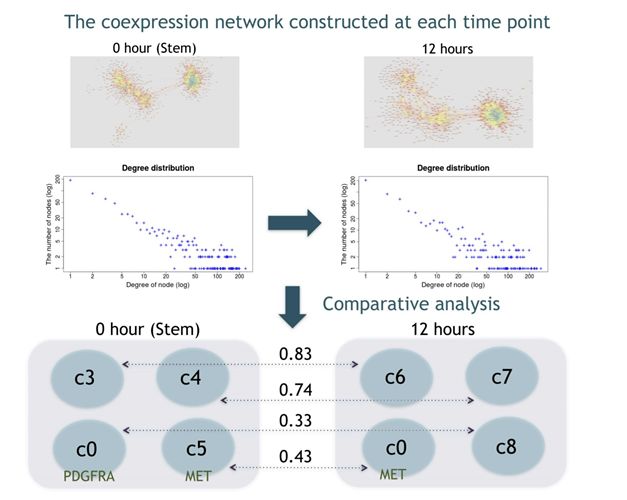
図: 疎なscRNA-seqデータからの遺伝子ネットワーク推定およびネットワーク比較
シングルセル解析を用いた種々の情報解析
ゲノム情報を1細胞レベルで観測するシングルセル解析は、生体組織や腫瘍組織に内在する細胞不均一性 (heteregeneity) や、細胞分化における状態遷移 (trajectory) 、確率的な遺伝子発現ゆらぎ (stochasticity) を推定する目的において用いられます。我々は主にシングルセル遺伝子発現量データ(scRNA-seq)を用いて、以下のようなプロジェクトに取り組んでいます。
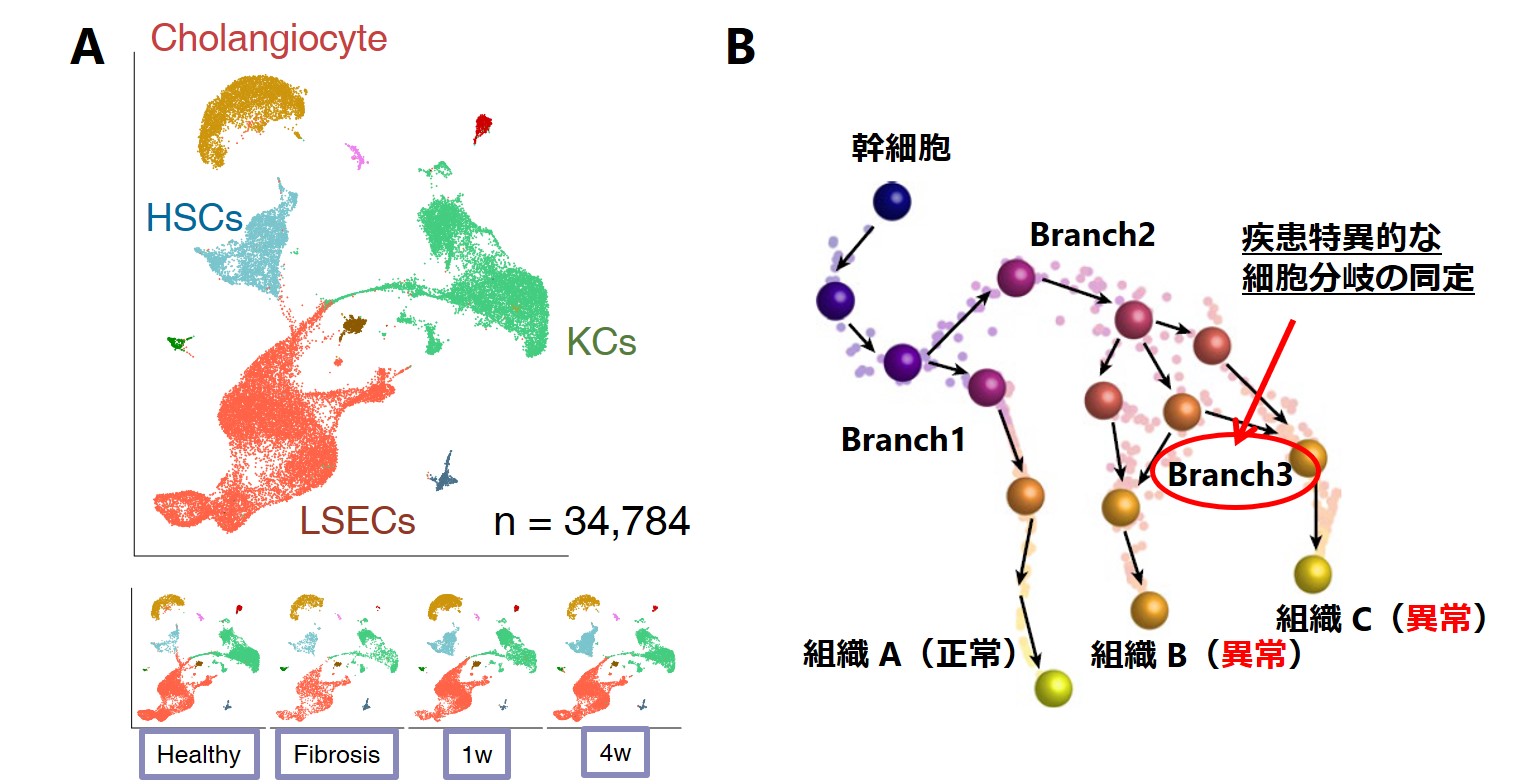
図: A: 肝細胞線維化の発症・治癒までを捉えた時系列解析。B: 幹細胞分化系とシングルセル解析を組み合わせた細胞分化軌道解析による細胞運命制御の調査。
我々は新学術領域「細胞ダイバース」において、シングルセル解析を柔軟かつ多面的に解析可能な1細胞解析プラットフォーム ShortCakeを構築しました(図5)。ShortCakeはDockerという技術を用いて解析環境そのものを「イメージ」としてダウンロード・共有可能なシステムであり、多くの研究者にとって最初の大きな障壁となるツールインストールのコストを大幅に削減できる仕組みとなっています。
細胞ダイバースの中で本ツールを利用して開催した1細胞解析技術講習会は好評を博しました。本講習のために作成した講習会資料は以下のWebサイトで公開しています。
・1細胞解析技術講習会資料
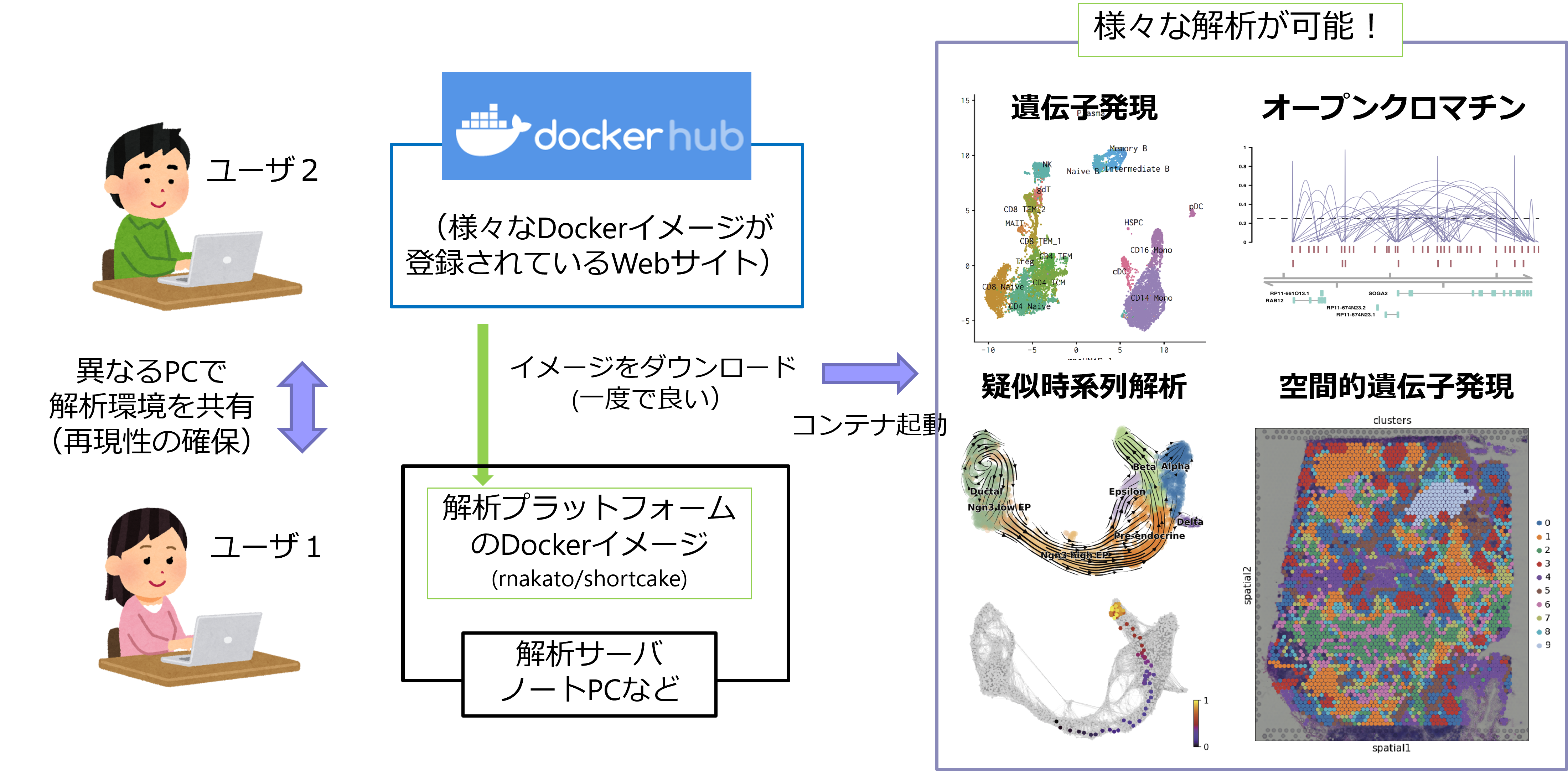
図: 1細胞解析プラットフォームShortCakeの概要
国際ヒトエピゲノムコンソーシアム (IHEC) プロジェクト (phase II)
我々は大規模エピゲノムデータ統合解析のためのアプローチを議論する国際ヒトエピゲノムコンソーシアム (IHEC)に参加しており、世界各国の情報解析の専門家と交流・議論しています。

2023年にカナダのバンフで開催されたIHEC年会での集合写真です。
IHECプロジェクトの一環として、国内外の多数の研究者との連携のもと、心臓・肺など9つの部位の血管内皮細胞を対象にエピゲノムデータ・遺伝子発現データを収集し、5テラバイトを超える大規模なエピゲノムデータベースを構築しました。
この大規模データセットから信頼度高く情報を抽出するため、個人差や技術的な要因によるデータ内ノイズを除去する様々な工夫や正規化手法を取り入れ、ノイズに対して頑健な解析ワークフローを構築しました。その結果、血管内皮細胞だけに存在する重要なゲノム機能部位(エンハンサーなど)を多数同定しました。さらなる解析により、血管内皮の性質は上半身と下半身で大きく分かれること、その多様性には特に遺伝子の転写制御に働くホメオボックス遺伝子群が強く寄与していること、これらの遺伝子群の発現制御にはゲノムの立体構造の変化が関わっていることなどが明らかになりました。
・ 東京大学プレスリリース
・ ヒト血管内皮エピゲノムデータベース
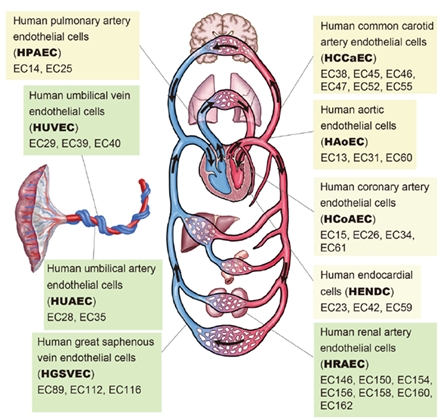
図: 全身をめぐる血管系の模式図
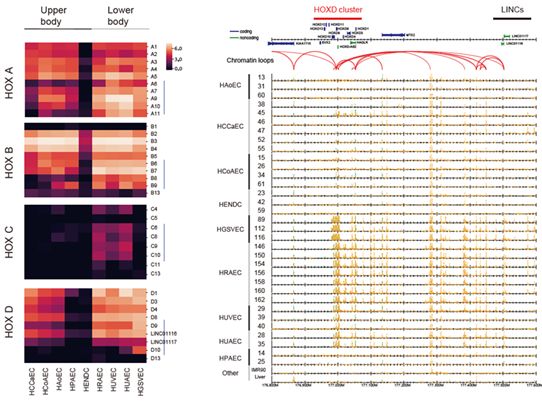
図: 左: 9部位の血管でのホメオボックス遺伝子の発現量を色の明るさで示したもの。部位ごとに発現量が大きく異なっていることがわかる。右: HOXDクラスター周辺のエンハンサーマーカーH3K27acの分布。赤い弧はChIA-PET解析で得られたクロマチンループ。
その他のツール
Hi-Cデータから多種多様な一次元特徴情報を効率的に抽出可能な新規手法 “HiC1Dmetrics”を開発しました。本手法ではHi-C解析において用いられる種々の既存指標を統一的に計算できるほか、これまで同定が難しかった特殊な立体構造(クロマチンハブ等)を定量的に抽出できる新規指標を提案しました。
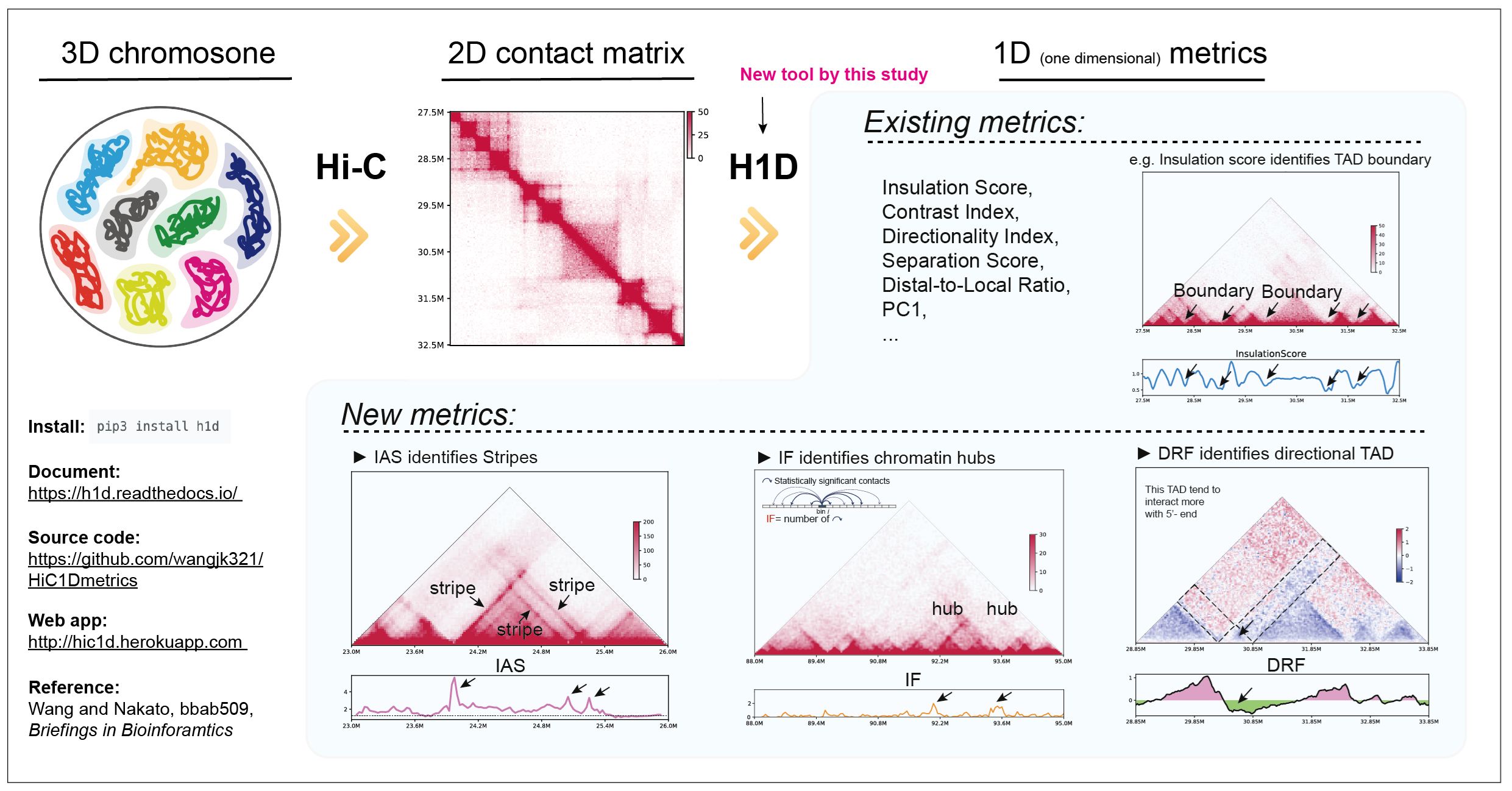
図: HiC1Dmetricsの概要
ChIP-seq解析はRNA-seqなどの他のオミクス情報とは異なり、ひとつのサンプルから複数のデータを生成する必要があるため(複数のヒストン修飾情報など)、サンプル数がしばしば多量になり、解析が煩雑になります。我々はそのような多数のChIP-seqデータを効率的に比較解析するためのパイプラインツール DROMPAplus を開発しました。DROMPAplusは種々の品質評価、フラグメント長推定、PCRバイアスのフィルタ、正規化、ピーク抽出、可視化やその他の解析に用いることができ、わかりやすいPDF形式で結果を出力します。
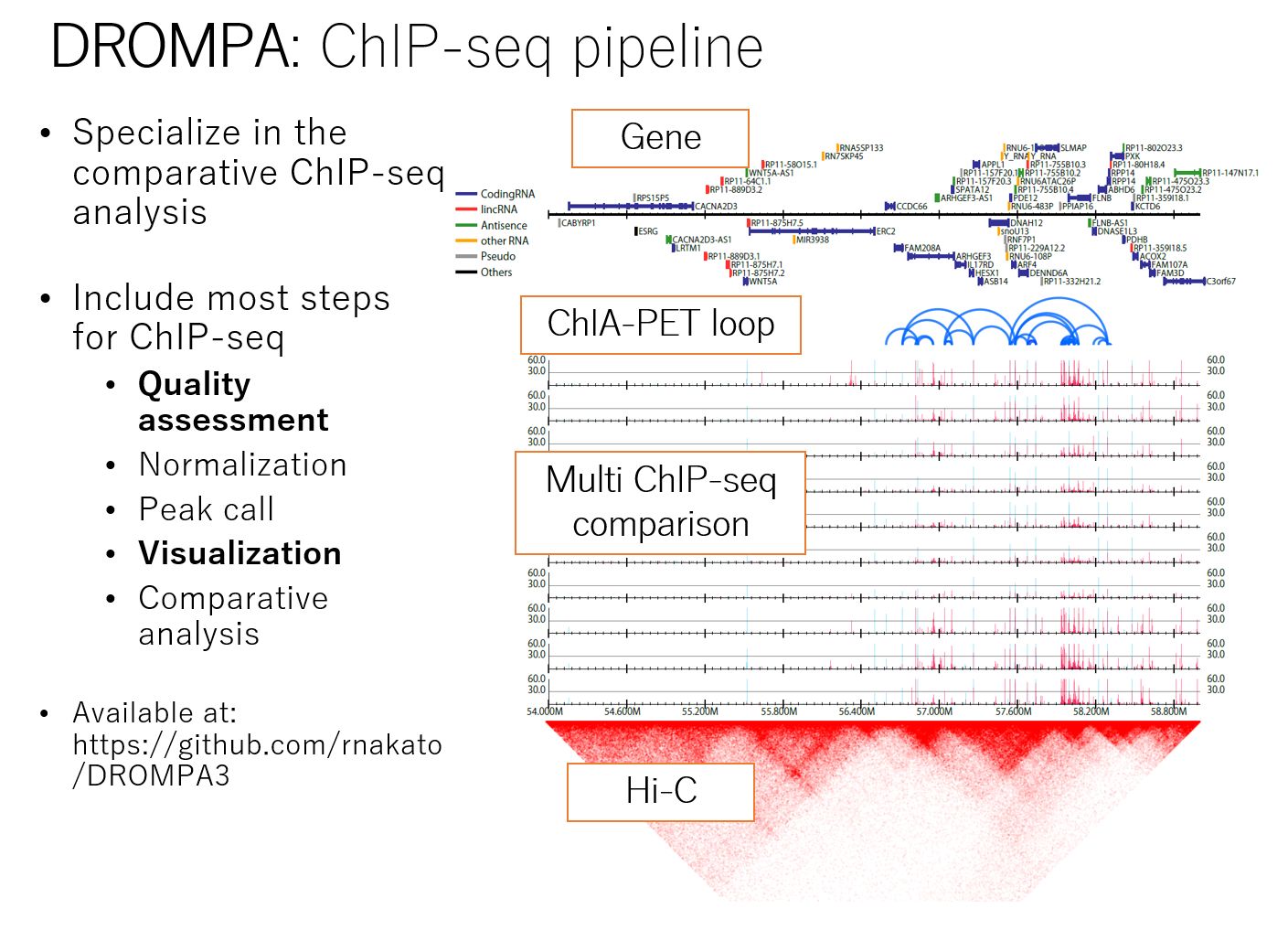
図: DROMPAplusの概要
大規模なNGS解析においては入力サンプルの品質が極めて重要です。多数のサンプルを自動的・客観的に品質評価するため、様々な指標がこれまで提案されています。しかし上記の血管内皮エピゲノムプロジェクトにおいて、推奨されているすべての品質評価基準を用いても除去しきれないある種の低品質サンプルがあることが明らかになりました。
そこで我々はChIP-seq解析のための新しい品質評価ツールSSPを開発しました。SSPは、鋭いピーク (point-source) 、広域に薄く分布するピーク (broad-source) 両方について、定量的で感度の高いS/N比を検出可能です。さらに、SSPは「得られたピークの信頼性」を評価する新しいスコアも提供しています。これにより、従来の品質評価基準で検出できなかった低品質サンプルを検出できるようになりました。これらのスコアは細胞種やリード数に依存しないため、様々な細胞種(または生物種)を含む多数サンプルの同時比較を可能にしています。
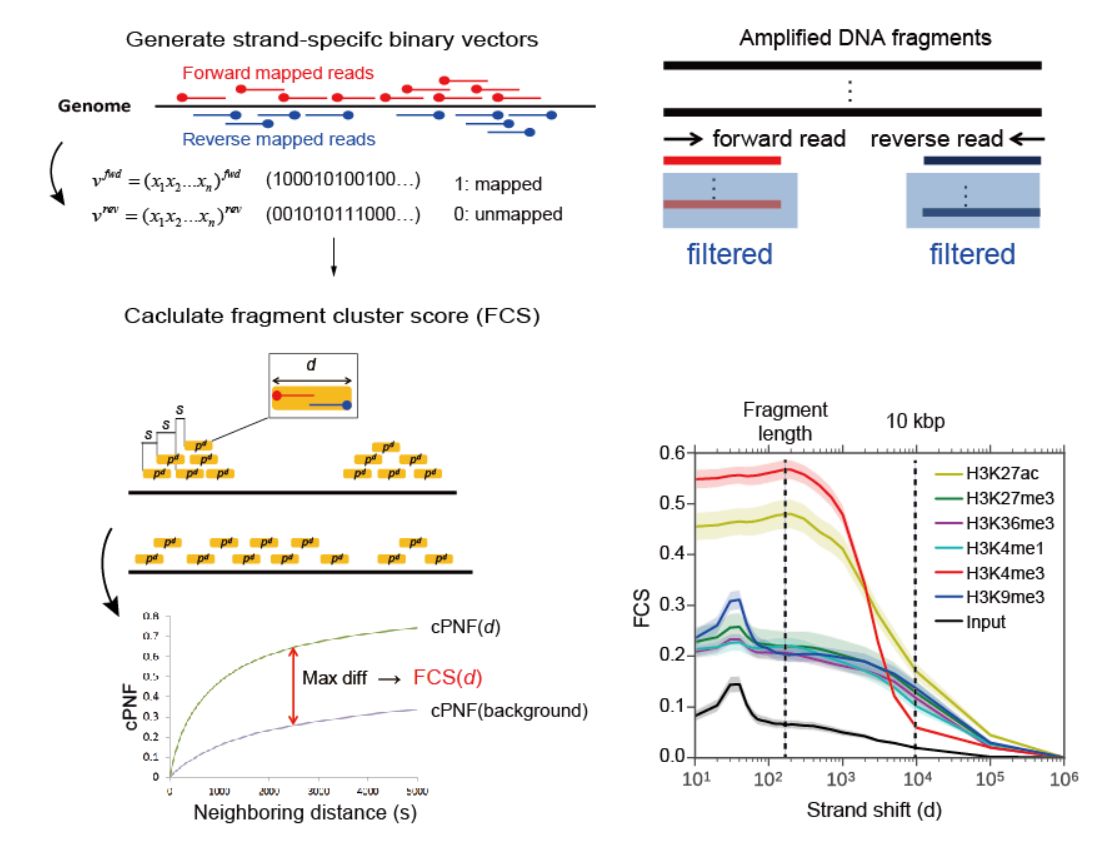
図: SSPの概要